|
聚类又称非监督分类,是一种探索性数据分析(Exploratory Data Analysis, EDA)方法,其目的是把有限数量的事物划分到若干“自然”的类别中。芯片结果的聚类分析很复杂,因为聚类本身就是个复杂的问题,聚类后的结果解析则更复杂。
在芯片数据分析中为什么要进行基因的聚类分析?主要是为了了解未知基因的功能。通过聚类分析我们把基因按某些特性(表达规律)分成若干类别,聚合成同一类的基因在表达上有相似规律,它们很可能具有某些类似的功能。基于这一假设,我们可以通过了解某一类别中已知功能的基因进一步分析其他未知基因的功能。
聚类分析方法(算法)大体上可分为层次聚类(hierarchical clustering)和分配聚类(partitional clustering)两种,其中层次聚类在芯片分析中用得比较多。经典的层次聚类也有两种:凝聚式层次聚类法(agglomerative hierarchical clustering)和分裂式层次聚类(divisive hierarchical clustering)。凝聚式层次聚类法先把N个基因当成N个独立的簇(或称群、类),通过逐个合并最相似的项最终归为一个簇;而分裂式层次聚类正好相反。
R语言发展到今天,研究者为聚类分析软件包的开发做出了大量的贡献(衷心地感谢他们),使用者有很多选择。读者可以从CRAN的Task view页面做进一步的了解。
1 计算距离:
先读入我们前面保存的分析数据:
data.fc <- read.csv("results.lim.all.csv", header=T, row.names=1)
head(data.fc, 2)
## T1h T24h T7d
## 254818_at 0.3409 6.024 6.215
## 245998_at -0.1367 3.676 2.778
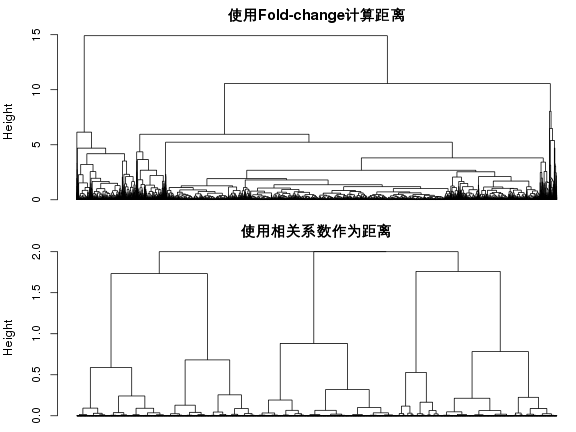
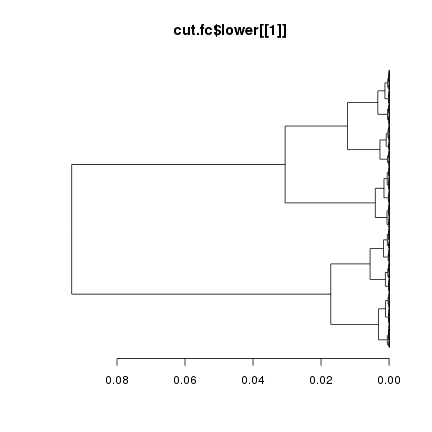
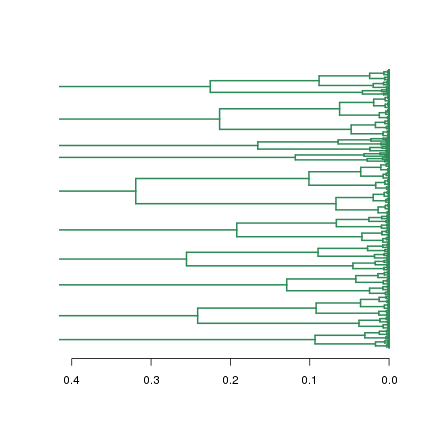
R中计算距离的函数为dist( )。但是使用什么指标来计算距离呢?一般通过计算相关系数作为基因间距离,R函数是cor( )。使用fold change计算距离也可以做出图来,其结果显然和用相关系数做出的图不一样:
par(mfrow=c(2,1))
par(mar=c(0,4,2,0))
dist.fc <- dist(data.fc, method="euclidean")
plot(hclust(dist.fc), hang=-1, labels=FALSE, main="使用Fold-change计算距离", sub="")
dist.fc <- as.dist(1 - cor(t(data.fc)))
plot(hclust(dist.fc), hang=-1, labels=FALSE, main="使用相关系数作为距离", sub="")
cor函数计算相关系数是计算矩阵不同列的相关系数,上面代码中函数 t() 对矩阵行列进行了转置,这样计算出的是基因间的相关系数矩阵。设置cor函数的method参数可以计算出pearson, kendall 或 spearman相关系数矩阵,默认计算pearson相关系数。as.dist函数把该矩阵转成了距离(向量)。如果直接使用dist函数计算距离,method可以设置距离计算的类型。
2 聚类
上面使用的hclust函数是R基本包中的凝聚式层次聚类函数。分裂式层次聚类运算耗时很长,做高通量数据分析一般不采用。
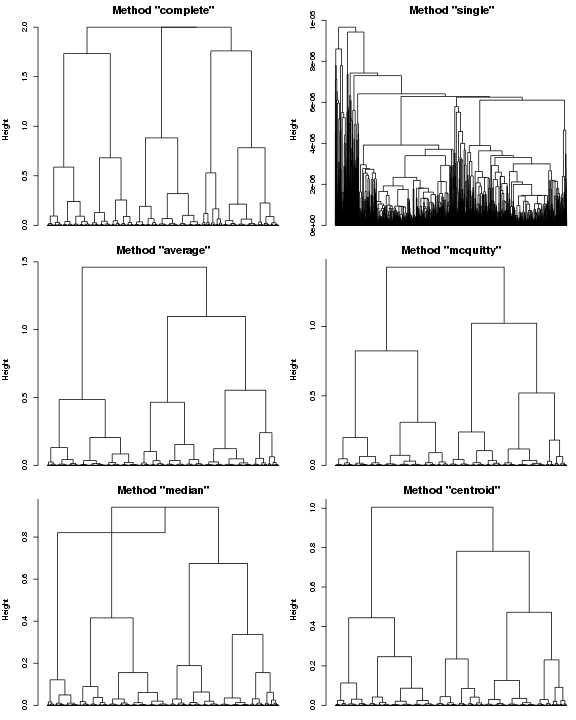
凝聚式层次聚类方法首先把N个基因当成N个独立的簇,每合并出一个新簇就为它计算相似系数。那么这个新簇的系数怎么计算?比如Ci和Cj合并后产生Cij,Cij在相似矩阵里面取什么值?这不是生物学研究者该考虑的问题,我们只用现成的。R的hclust函数提供了7中可用方法,"ward", "single", "complete", "average", "mcquitty", "median" 和 "centroid"。
我们看看其中的6种结果:
par(mfrow=c(3,2))
par(mar=c(0,4,2,0))
plot(hclust(dist.fc, method="complete"), hang=-1, labels=FALSE, main='Method "complete"', sub="")
plot(hclust(dist.fc, method="single"), hang=-1, labels=FALSE, main='Method "single"', sub="")
plot(hclust(dist.fc, method="average"), hang=-1, labels=FALSE, main='Method "average"', sub="")
plot(hclust(dist.fc, method="mcquitty"), hang=-1, labels=FALSE, main='Method "mcquitty"', sub="")
plot(hclust(dist.fc, method="median"), hang=-1, labels=FALSE, main='Method "median"', sub="")
plot(hclust(dist.fc, method="centroid"), hang=-1, labels=FALSE, main='Method "centroid"', sub="")
更多的聚类方法和函数请参看 CRAN Task View。
3 系统树分枝截取和簇定义
聚类分析的目的是为了获得合适的分类。但怎么才算“合适”?这也是个大问题。典型的做法是先设定一个阈值,再使用这个阈值截取系统树,能截出几个系统树分枝就是几个簇。通过hclust类型数据截取系统树分枝分3步进行:
- 用cutree函数截取hclust数据
- 获取各分枝的基因名称并通过基因名称取距离矩阵的子集
- 使用距离矩阵子集重构系统树
后两步都很熟悉了,主要看看第1步。cutree函数参数h是我们设置的截取阈值,得到的数据是带名称的向量:元素名称是基因名,元素的值是系统树分枝编号。
clust.fc <- hclust(dist.fc, method="complete")
branches.fc <- cutree(clust.fc, h=0.4)
#看看数据头的样子
head(branches.fc)
## 254818_at 245998_at 265119_at 256114_at 254805_at 251775_s_at
## 1 2 2 2 1 2
#截取出的分枝和各分枝基因数
table(branches.fc)
## branches.fc
## 1 2 3 4 5 6 7 8 9 10
## 1960 2415 3035 845 592 1410 1655 1459 1574 1060
接下来可以取距离矩阵子集并重构第系统树分枝。 你还可以在原系统树上通过画方框的方法标出各分枝:
br1 <- names(branches.fc[branches.fc==1])
dist.br1 <- as.dist(as.matrix(dist.fc)[br1,br1])
clust.br1 <- hclust(dist.br1, method="complete")
br3 <- names(branches.fc[branches.fc==3])
dist.br3 <- as.dist(as.matrix(dist.fc)[br3,br3])
clust.br3 <- hclust(dist.br3, method="complete")
par(mfrow=c(1,3))
par(mar=c(1,4,2,0))
plot(clust.br1, hang=-1, labels=FALSE, main='branch 1', sub="")
plot(clust.br3, hang=-1, labels=FALSE, main='branch 3', sub="")
plot(clust.fc, hang=-1, labels=FALSE, main='clust.fc', sub="")
rect.hclust(clust.fc, h=0.4, border="red")
但从上图框出的分枝大小和前面的分枝基因数来看,各分枝并不是按图上横坐标的顺序进行排列的。
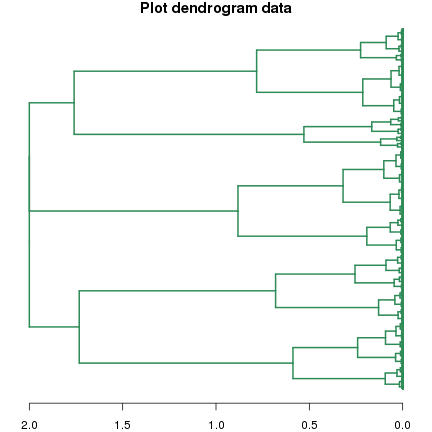
如果你的目的是获得基因列表,那么上面的方法就可以了;如果想获得更加美观点的图形,可以将hclust产生的结果转为系统树(dendrogram)类型。系统树类型的数据也可以直接使用plot函数绘图,参数比hclust绘图更多:
dend.fc <- as.dendrogram(clust.fc, hang=-1)
par(mfrow=c(1,1))
par(mar=c(2,1,1,1))
plot(dend.fc, leaflab="none", horiz=T, edgePar = list(col="seagreen", lwd=2), main="Plot dendrogram data")
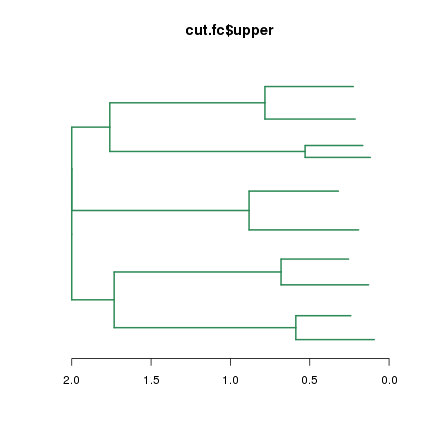
而且还可以使用cut函数截取系统树。cut( )函数通过设定height参数(阈值)来截取系统树,获得的结果是列表结构,有名称为upper和lower的两个列表成员:
cut.fc <- cut(dend.fc, h=0.4)
class(cut.fc)
再看upper和lower的类型,发现upper是系统树类型,而lower是列表。把upper系统树plot处理你就知道它是什么了:
class(cut.fc$upper); class(cut.fc$lower);
plot(cut.fc$upper, leaflab="none", horiz=T, edgePar = list(col="seagreen", lwd=2), main="cut.fc$upper")
再看看lower列表,它有10个列表元素,每个都是系统树数据,我们当然也可以plot这些系统树分枝:
cnames <- NULL
for(i in 1:length(cut.fc$lower)) cnames <- c(cnames, (class(cut.fc$lower[[i]])))
cnames
## [1] "dendrogram" "dendrogram" "dendrogram" "dendrogram" "dendrogram"
## [6] "dendrogram" "dendrogram" "dendrogram" "dendrogram" "dendrogram"
plot(cut.fc$lower[[1]],leaflab="none", horiz=T, main="cut.fc$lower[[1]]")
事实上我们可以通过另外的方法看到这10个分枝:
plot(dend.fc, leaflab="none", horiz=T, edgePar = list(col="seagreen", lwd=2), xlim=c(0.4,0))
请看cut.fc$lower1是不是就是最下面的分支?从这也可以看出cut.fc$lower中各项的排列顺序,接下来再分析各个分支就有谱了。dendrogram类型数据要获得基因名称列表还需返回到hclust数据类型,然后通过hclust的labels属性获得基因列表。因为cutree和cut函数产生的分枝编号不能对应起来,这一步很必要。
clust.br1 <- as.hclust(cut.fc$lower[[1]])
genes.br1 <- clust.br1$labels
4 基因列表分析
基因列表的GO和KEGG分析我们前面已经说过了,这里主要看看各簇(1-9簇)基因的表达变化趋势:
data.fc <- as.matrix(data.fc)
par(mfrow=c(3,3))
par(mar=c(2,3,2,1))
par(mgp=c(2, 0.5, 0))
library(scales)
color <- alpha("blue", alpha=0.1)
for(i in 1:9){
genes <- as.hclust(cut.fc$lower[[i]])$labels
exprs <- data.fc[genes,]
ylim = c(min(exprs), max(exprs))
plot(as.vector(exprs[1,]), col=color, type='l', ylim=ylim, ylab="Expression( LogFC)", main=paste("Cluster", i))
for(j in 2:nrow(exprs)) points(as.vector(exprs[j,]), col=color, type='l')
box()
}
6 Session Info
## R version 3.1.0 (2014-04-10)
## Platform: x86_64-pc-linux-gnu (64-bit)
##
## locale:
## [1] LC_CTYPE=zh_CN.UTF-8 LC_NUMERIC=C
## [3] LC_TIME=zh_CN.UTF-8 LC_COLLATE=zh_CN.UTF-8
## [5] LC_MONETARY=zh_CN.UTF-8 LC_MESSAGES=zh_CN.UTF-8
## [7] LC_PAPER=zh_CN.UTF-8 LC_NAME=C
## [9] LC_ADDRESS=C LC_TELEPHONE=C
## [11] LC_MEASUREMENT=zh_CN.UTF-8 LC_IDENTIFICATION=C
##
## attached base packages:
## [1] tcltk stats graphics grDevices utils datasets methods
## [8] base
##
## other attached packages:
## [1] scales_0.2.4 zblog_0.1.0 knitr_1.5
##
## loaded via a namespace (and not attached):
## [1] colorspace_1.2-4 evaluate_0.5.3 formatR_0.10 highr_0.3
## [5] munsell_0.4.2 plyr_1.8.1 Rcpp_0.11.1 stringr_0.6.2
## [9] tools_3.1.0
---------------------
本文来自 金子哦 的CSDN 博客 ,全文地址请点击:https://blog.csdn.net/u014801157/article/details/24372399?utm_source=copy
|